
Dynamic Homeostasis
Metabolic Stability through Coordinated Energy Investment
Living cells persist in a state of dynamic homeostasis far from thermodynamic equilibrium. At this foundational phase of metabolic regulation, the cell does not passively maintain order but continuously invests energy to preserve biochemical gradients, redox balance, and structural coherence. The biochemical infrastructure of this phase integrates oxidative phosphorylation with substrate-level pathways and regulatory feedback loops that enable a stable, entropy-exporting system.
1. State: Energetic Coherence through Oxidative Metabolism
Under resting, oxygen-rich conditions, ATP demand is primarily met through oxidative phosphorylation (OXPHOS), which generates more than 90% of cellular ATP. This is complemented by cytosolic glycolysis, modest glycogen turnover, background fatty acid oxidation, and active flux through the tricarboxylic acid (TCA) cycle. Together, these pathways stabilize intracellular energy levels, minimize entropy accumulation, and buffer against transient metabolic fluctuations.
- ATP levels: High, with a low AMP/ATP ratio (~0.01)
- ΔG (ATP hydrolysis): ~–55 to –60 kJ/mol
- Mitochondrial membrane potential (Δψ): –150 to –180 mV
- Redox state: Balanced NAD⁺/NADH and FAD/FADH₂ pools
- ROS levels: Low and buffered by SOD, glutathione, catalase
2. Key Systems: Biochemical Pathways in Energetic Harmony
a) Oxidative Phosphorylation: Mitochondrial Core Engine
Electrons from NADH and FADH₂, generated by glycolysis, β-oxidation, and the TCA cycle, flow through the mitochondrial electron transport chain (ETC), pumping protons and generating a proton motive force (Δψ + ΔpH) to drive ATP synthase.
Reactions:
- NADH + H⁺ + ½ O₂ → NAD⁺ + H₂O (ΔG°’ ≈ –220 kJ/mol)
- ADP + Pi → ATP (ΔG°’ ≈ +30.5 kJ/mol)
Net yield:
- 3 ATP/NADH
- 2 ATP/FADH₂
ATP is regenerated at a pace tuned to membrane potential and ADP availability.
b) Glycolysis – Cytosolic Gateway and Modulator
Glycolysis converts glucose → 2 pyruvate + 2 NADH + 2 ATP. It functions as a flexible gatekeeper, matching substrate inflow and ATP output.
Control enzymes:
- Hexokinase (irreversible): glucose → glucose-6-P
- PFK-1: fructose-6-P → fructose-1,6-bisP (main control point, AMP-sensitive)
- Pyruvate kinase: phosphoenolpyruvate → pyruvate
Regulation:
- Inhibited by ATP, citrate
- Activated by AMP, fructose-2,6-bisP (PFKFB3-dependent)
c) Glycogen Metabolism – Short-Term Buffering
Glycogen serves as a glucose reserve. In muscle and liver, glycogen phosphorylase breaks it down into glucose-1-phosphate → glycolysis.
- Activated by AMP, Ca²⁺, epinephrine
- Inhibited by ATP, glucose-6-phosphate
- Synthase (insulin-driven) promotes glycogenesis during energy abundance
This mechanism buffers energy without immediate dependence on blood glucose.
d) Fatty Acid Oxiation – Baseline Lipid Use
Even in glucose-dominant states, β-oxidation of fatty acids occurs at a low basal rate to reduce glucose pressure:
- FA-CoA transported via carnitine shuttle
- β-oxidation yields acetyl-CoA, NADH, FADH₂
- Acetyl-CoA enters TCA; cofactors support ETC
e) Tricarboxylic Acid (TCA) Cycle – Substrate Convergence Hub
This mitochondrial cycle integrates carbohydrate (pyruvate), lipid (acetyl-CoA), and amino acid catabolism.
Key reactions:
- Pyruvate + NAD⁺ + CoA → Acetyl-CoA + CO₂ + NADH
- 1 turn: 3 NADH, 1 FADH₂, 1 GTP, 2 CO₂
Regulation:
- Isocitrate dehydrogenase, α-ketoglutarate dehydrogenase: activated by ADP, Ca²⁺; inhibited by NADH
3. Physiology: Flexibility without Emergency Activation
- High glucose availability allows OXPHOS to dominate
- Glycolysis adjusts rapidly to ATP spikes (e.g., muscle contraction)
- Glycogen buffers fasting or delayed intake
- Lipid oxidation supports low-glucose tissues (e.g., heart, liver)
- Cofactor cycling (NAD⁺/NADH) ensures redox balance across pathways
Summary:
Dynamic homeostasis in Phase 1 is not about stasis—it is about coherence across temporally integrated, thermodynamically tuned metabolic subsystems. Oxidative phosphorylation is the dominant source of ATP, but only through the orchestration of glycolysis, glycogen turnover, TCA flux, and low-level lipid oxidation can the cell sustain low entropy, directional flux, and energy sufficiency.
This phase reflects not only a thermodynamic minimum of entropy production but also a maximally efficient configuration of energy transformation—a poised system, energetically ready to adapt, resist, or reorganize.
Disruption
Disruption is the first systemic deviation from metabolic homeostasis. It begins when oxidative phosphorylation can no longer meet ATP demand, or when substrate availability, oxygen supply, or redox balance becomes insufficient to maintain the energy gradients that preserve structural and biochemical order.
This is not merely a metabolic slowdown—it is a thermodynamic crisis: the system continues to consume ATP while failing to regenerate it at sustainable rates, leading to increased entropy and biochemical instability.
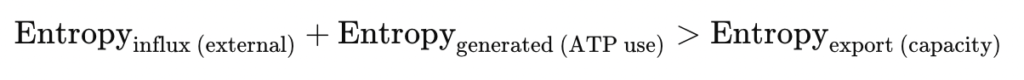
Common Triggers of Mitochondrial Disruption: Heat stress, Hypoxia, Acidosis, Oxidative or inflammatory stress
| Trigger Type | Description |
|---|---|
| Hypoxia | ↓ O₂ availability halts ETC Complex IV → ↓ proton motive force (Δψ), ATP synthesis fails. |
| Mitochondrial Dysfunction | ROS damage, mtDNA mutations, or loss of membrane potential impairs ETC efficiency. |
| Excess ATP Demand | Sudden increases in ATP use (e.g., rapid signaling, contraction, immune response) outpace oxidative regeneration. |
| Redox Imbalance | Accumulated NADH and FADH₂ (due to stalled ETC) inhibit upstream reactions (e.g., glycolysis, TCA). |
| Substrate Limitation | Exhaustion of glucose, glycogen, or fatty acids impairs acetyl-CoA and NADH production. |
| Lactic Acidosis | Anaerobic glycolysis leads to lactate buildup → ↓ pH → inhibition of metabolic enzymes. |
2. Biochemical Breakdown Events
a) Collapse of Oxidative Phosphorylation
- ETC flux halts → Δψ and ΔpH dissipate
- ATP synthase (Complex V) reverses direction (hydrolyzing ATP instead of synthesizing it)
- NAD⁺/NADH and FAD/FADH₂ ratios invert → metabolic traffic jams
- ↑ ROS from Complex I/III electron leakage → damages lipids, proteins, and mtDNA
b) Accumulation of Metabolic Intermediates
- Pyruvate builds up → converted to lactate via lactate dehydrogenase
Pyruvate + NADH ↔ Lactate + NAD⁺ - ↓ NAD⁺ stalls glycolysis (at glyceraldehyde-3-phosphate dehydrogenase)
- Citrate, succinate, and acetyl-CoA accumulate → feedback inhibition on upstream flux
- AMP levels rise → AMPK activation threshold nears
c) Energetic Signals of Disruption
- AMP/ATP ratio increases → activates sensing pathways (AMPK, mTOR inhibition)
- ΔG of ATP hydrolysis becomes less negative → ATP is less efficient as an energy source
- Glycogenolysis is triggered, but glycogen stores are finite
- Fatty acid β-oxidation cannot compensate fully without oxygen, increasing ROS burden
3. Functional Consequences:
a) Metabolic Instability
- ATP hydrolysis continues: enzymes, transporters, and cytoskeletal processes persist
- ATP synthesis stalls: regeneration via mitochondria is compromised
- Net ΔS (entropy) increases as chemical gradients flatten
b) Loss of Hierarchical Control
- Pathways desynchronize: glycolysis accelerates, but pyruvate is trapped as lactate
- Feedback inhibition fails: citrate builds up, but can’t modulate glycolysis effectively
- Lipid catabolism increases, but without proper oxidation, fatty acid intermediates accumulate (e.g., acyl-CoA, ketone bodies)
c) Cellular Events
- Mitochondrial swelling, loss of cristae structure
- Organelle mislocalization (due to ATP-dependent transport failure)
- pH drops, impairing enzyme kinetics
- Ion gradients collapse → Na⁺/K⁺-ATPase and Ca²⁺-ATPase slow, leading to osmotic and signaling stress
- Cytoskeletal disruption → loss of intracellular polarity
4. Transition to Phase 3:
Disruption sets the thermodynamic preconditions for Reaction:
- ATP is depleted
- Entropy rises
- Structural order deteriorates
But containment has not yet begun. This phase ends when the metabolic system crosses threshold signals (AMPK, ROS, Ca²⁺ spikes) and initiates active defense and reprogramming responses, launching Phase 3: Reaction.
Summary:
Phase 2 is defined not by passive metabolic decline, but by active decoupling: ATP consumption continues, but generation fails to keep pace. Oxidative phosphorylation falters, glycolysis accelerates anaerobically, and redox feedbacks lose coherence. The cell enters a state of net energy loss, initiating a cascade of biochemical noise, oxidative damage, and structural unraveling.
This is not yet the adaptive response. It is the thermodynamic exposure of vulnerability—a fall into metabolic instability that marks the need for emergency containment.
Reaction
Following disruption, the metabolic system enters a crisis state. Oxidative phosphorylation is stalled or insufficient, ATP is depleting, and redox and ion imbalances are escalating. Reaction is the phase where containment overrides optimization. The cell rapidly reallocates remaining resources to stabilize key variables (ATP, NAD⁺, ROS) and maintain basic functionality.
This is not recovery — it is triage: a short-term, high-cost operation to delay collapse.
| Signal | Function |
|---|---|
| AMP/ATP ratio ↑ | Activates AMPK → inhibits anabolic processes, enhances catabolism |
| NADH/NAD⁺ imbalance | Activates lactate dehydrogenase → regenerates NAD⁺ for glycolysis |
| ROS accumulation | Triggers Nrf2 and antioxidant response systems |
| Cytosolic Ca²⁺ spikes | Initiate membrane repair, cytoskeletal reorganization, and UPR (unfolded protein response) |
| Lactate build-up / pH drop | Inhibits sensitive enzymes; signals glycolytic reliance |
a) Glycolytic Acceleration
The ETC is compromised, so the cell relies on anaerobic glycolysis for rapid, short-term ATP:
- Glucose → 2 Pyruvate → 2 Lactate
- Net gain: 2 ATP per glucose
- NAD⁺ regenerated via lactate dehydrogenase
- Upregulated by AMPK, HIF-1α, and PFKFB3
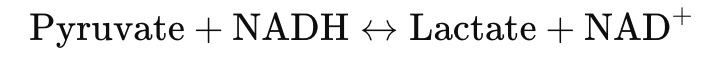
b) Activation of AMPK
AMP-activated protein kinase (AMPK) becomes the master regulator of crisis metabolism:
- Inhibits:
- mTOR → halts protein synthesis
- Acetyl-CoA carboxylase (ACC) → suppresses fatty acid synthesis
- Activates:
- PFK-2 → enhances glycolytic flux
- GLUT1/4 → boosts glucose uptake
- Autophagy → clears damaged mitochondria and recycles nutrients
AMPK shifts the cell into a high-efficiency, energy-sparing mode.
c) Antioxidant Defense
Oxidative stress rises as ETC dysfunction leads to ROS burst.
- Nrf2 pathway activates transcription of:
- SOD (superoxide dismutase)
- Catalase
- Glutathione peroxidase
- Goal: contain oxidative entropy and prevent irreversible damage to DNA, proteins, and membranes
d) Lactate Export
To prevent lactic acidosis:
- Monocarboxylate transporters (MCT1/4) shuttle lactate + H⁺ out of the cell
- Reduces intracellular acid load
- Temporarily maintains cytosolic pH and redox homeostasis
e) Autophagy & Proteostasis Buffering
Stress-activated autophagy allows degradation of dysfunctional organelles and proteins:
- Removes damaged mitochondria (mitophagy)
- Recycles amino acids and lipids
- Stabilizes intracellular composition under ATP-limited conditions
Concurrently:
- Heat Shock Proteins (HSPs) attempt to refold misfolded proteins
- UPR downregulates protein synthesis and enhances ER-associated degradation (ERAD)
Substrate and Enzym Shift:
| Pathway | Change |
|---|---|
| Glycolysis | ↑↑ (PFK-1 activated, pyruvate kinase supported) |
| TCA cycle | ↓ (substrate accumulation + NADH inhibition) |
| ETC | ↓↓↓ (Δψ collapse, Complex IV bottleneck) |
| β-oxidation | Mixed: ↑ in tissues with residual oxygen, but ↓ in hypoxia |
| Lipid synthesis | ↓ (AMPK inhibition of ACC) |
| Glycogenolysis | ↑ (glucagon/epinephrine-driven release) |
| Gluconeogenesis | ↓ (energetically expensive, suppressed under AMPK control) |
Thermodynamic Characteristics
- ATP hydrolysis continues, but regeneration is low → net ΔG becomes less negative
- Entropy production rises: lactate, ROS, unfolded proteins, ion gradients lost
- Δψ, ΔpH, and NAD⁺ levels are buffered but unstable
- The system teeters on collapse: energy demand remains high, but substrate and redox capacity are diminished
Summary: Reaction is:
- High-cost, low-efficiency, but absolutely necessary
- Not sustainable, but buys time for possible reorganization
- Defined by:
- Lactate-driven ATP generation
- AMP/ROS-triggered defensive pathways
- Suppression of growth and synthesis
If these containment systems succeed, the cell transitions to Phase 4: Adaptation. If not, it progresses toward apoptosis or necrosis.
Adaptation
While Phase 3 (Reaction) contains collapse, it cannot restore systemic function. Adaptation begins once ATP levels, redox balance, and ion gradients are partially stabilized. It is a cell-wide realignment—of metabolic priorities, enzyme expression, and structural allocation—designed not to return to the previous state, but to build a new one optimized for the post-crisis condition.
Adaptation relies on functional mitochondrial recovery, enhanced metabolic flexibility, and transcriptional reprogramming.
1.Restoration of Mitochondrial Function
The core thermodynamic engine—oxidative phosphorylation (OXPHOS)—must be reactivated:
- ETC Complex repair: dysfunctional proteins removed via mitophagy; new complexes synthesized
- Membrane potential recovery (Δψ): proton gradients reestablished through ETC flow
- Redox balance restored: NAD⁺/NADH ratio returns to forward-driving levels
- Oxygen supply normalized or optimized via angiogenesis or hypoxia adaptation
This re-couples ATP hydrolysis with ATP regeneration (ΔG becomes more negative, ~–55 kJ/mol).
Dual Track ATP-Production:
| Pathway | Function |
|---|---|
| OXPHOS (mitochondrial) | High-yield ATP regeneration resumes via NADH and FADH₂ oxidation |
| Glycolysis (cytosolic) | Remains active in parallel to support rapid local energy needs and buffer oxygen variability |
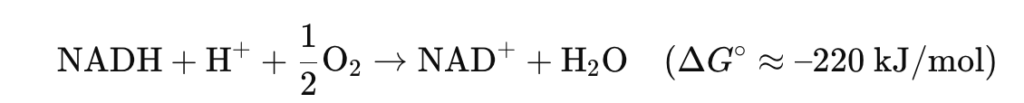
This hybrid model maintains metabolic flexibility and ensures survival under variable oxygen and substrate conditions.
3.Activation of Lipid Oxidation
Fatty acids become a key long-term energy substrate:
- AMPK lifts inhibition on carnitine palmitoyltransferase I (CPT1) → imports fatty acyl-CoA into mitochondria
- β-oxidation generates:
- Acetyl-CoA → fuels TCA cycle
- NADH / FADH₂ → feeds ETC
- Provides stable ATP without depleting limited glucose reserves
Key Equation:
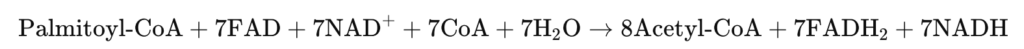
This transition signifies metabolic stabilization and redox-supported OXPHOS reengagement.
4. Transcriptional & Enzymatic Reprogramming
Cellular machinery is rewritten to sustain the new metabolic logic:
- PGC-1α and NRF1/2: drive mitochondrial biogenesis
- TFAM: boosts mitochondrial DNA transcription
- HIF-1α (if hypoxia persists): stabilizes expression of glycolytic enzymes and angiogenic factors
- Histone acetylation via Acetyl-CoA: links energetic state to epigenetic changes
Protein isoforms, enzyme levels, and even organelle distribution are now tuned to the adapted energy infrastructure.
5.Proteostasis and Quality Control
The cleanup from Phase 3 continues:
- Autophagy clears residual damage (e.g., oxidized proteins, lipid peroxides)
- Chaperones restore proteome order (HSP70, HSP90)
- Proteasome degrades improperly folded or aggregated proteins
These systems are downregulated gradually as stability returns.
Thermodynamic Markers of Stability:
| Metric | Adaptive State |
|---|---|
| ATP levels | Rising and sustainable |
| ΔG of ATP hydrolysis | More negative (~ –55 kJ/mol) |
| Δψ and ΔpH | Fully restored |
| AMP/ATP ratio | Approaching baseline |
| ROS levels | Controlled, with antioxidant defenses normalized |
| Entropy production | Declining and matched by entropy export |
Summary: Building a New Metabolic Identity
Adaptation is where the metabolic system restructures itself for endurance, not just survival:
- It stabilizes ATP supply chains through restored OXPHOS and parallel glycolysis.
- It reprioritizes lipid catabolism for long-term energy.
- It rewires gene expression to match new metabolic demands.
- It reduces entropy burden through efficient clearance and regeneration.
This is not a return to Phase 1. It is the construction of a new, viable metabolic regime—preparing the ground for the next phase: Refined Homeostasis.
Refined Homeostasis
Refined Homeostasis is not a return to pre-disruption metabolism. Instead, it reflects the integration of adaptations, the stabilization of energy flow, and the reprogramming of metabolic identity. The cell no longer responds to crisis but maintains a coherent internal environment, informed by past stress, optimized for current needs, and primed for future fluctuations.
This phase marks a shift from flexible resilience to stabilized precision.
1. ATP Regeneration Re-Coupled to ATP Demand
Energetic balance is re-established through the tight coupling of ATP synthesis and hydrolysis:
- OXPHOS is dominant, with high efficiency (~90% of ATP)
- Glycolysis remains active but context-specific (e.g., localized or signaling-driven)
- Fatty acid oxidation supports baseline ATP, particularly in liver, muscle, and heart
ΔG of ATP hydrolysis stabilizes around –55 to –60 kJ/mol, indicating optimal energy use.
The energy system operates like a closed loop: inputs (glucose, fatty acids, oxygen) are matched to the exact needs of the system, with minimal waste and maximal thermodynamic control.
Integrated Metabolic Networks:
Multiple fuel sources operate in harmony, each fulfilling specific roles:
| Pathway | Role in Refined Homeostasis |
|---|---|
| TCA Cycle | Central integrator of substrates (carbs, fats, amino acids) |
| β-Oxidation | Basal energy support and lipid turnover |
| Glycolysis | Rapid, local ATP generation and metabolic intermediates |
| Glycogen Metabolism | Dynamic glucose buffering and signal-dependent mobilization |
| Amino Acid Metabolism | Anaplerotic balance and stress-responsive flux |
| Pentose Phosphate Pathway | Maintains NADPH and biosynthesis |
These systems form a networked infrastructure, enabling adaptive control without invoking emergency responses.
3. Redox and Cofactor Homeostasis
NAD⁺/NADH and FAD/FADH₂ ratios are tightly controlled:
- Support steady electron delivery to ETC
- Maintain forward flux through glycolysis and TCA
- Prevent excessive ROS through balanced oxidation
Additionally:
- Acetyl-CoA levels are partitioned between the TCA cycle, lipid synthesis, and epigenetic regulation (e.g., histone acetylation)
- AMP/ATP ratio returns to basal (~0.01), keeping AMPK inactive but primed
Epigenetic Encoding and Transcriptional Memory
Adaptations from earlier phases are encoded into transcriptional programs:
- PGC-1α, HIF-1α, and AMPK targets are integrated into stable gene expression
- Chromatin is reorganized to reflect new energetic priorities
- Metabolic enzymes and transporters are rebalanced to match long-term functional needs
This metabolic memory enables faster and more efficient responses to future stressors.
Structural and Organelle Reorganization
Intracellular organization is optimized for spatial energy distribution:
- ATP microdomains are restored through organelle positioning and cytoskeletal anchoring
- Mitochondria are redistributed (e.g., near energy-demanding zones like synapses, immune synapses, or contractile zones)
- Membrane transporters and pumps (e.g., Na⁺/K⁺-ATPase, GLUT4) are re-tuned to the new demand landscape
Thermodynamic Characteristics:
| Parameter | Stabilized Value |
|---|---|
| ATP / ADP / AMP | High ATP, low AMP (homeostatic range) |
| Δψ (mitochondrial potential) | ~ –150 to –180 mV |
| ΔG (ATP hydrolysis) | ~ –55 to –60 kJ/mol |
| ROS levels | Minimal, with effective scavenging |
| Entropy export | Balanced with internal production |
| Enzyme activity | Tuned to demand; not maximal, but efficient |
Summary: Thermodynamic Coherence as Stability
In Refined Homeostasis, the metabolic infrastructure operates not at maximum capacity, but maximum coherence.
- Energy production meets—not exceeds—demand
- Substrate use is flexibly prioritized (glucose, fats, amino acids)
- Redox and cofactor cycling are optimized for stability and low noise
- Epigenetic and transcriptional memory encode past experience into present resilience
- The system is ready for change, but no longer in crisis
This phase represents a dynamic attractor—an energetic and informational architecture that integrates biochemical flux, structural organization, and past regulatory adaptation into a self-sustaining regime of cellular function.
Conclusion
Living cells do not maintain stability through static equilibria but through dynamically regulated metabolic cycles that resist entropy while adapting to internal and external perturbations. This framework conceptualizes metabolism as a five-phase thermodynamic process, where energy flow, redox balance, and substrate flexibility sustain order far from equilibrium.
Under oxygen-rich conditions, metabolism is anchored in oxidative phosphorylation, efficiently converting glucose (via glycolysis and the TCA cycle) into ATP through the electron transport chain. Complementary pathways—glycogen turnover, basal lipid oxidation, and redox cycling—sustain energetic balance and biochemical order. The system is poised, responsive, and low in entropy.
Disruption occurs when energy demand outpaces mitochondrial ATP regeneration—due to hypoxia, oxidative stress, or substrate limitation. Mitochondrial gradients collapse, NADH accumulates, and glycolysis shifts anaerobically to lactate. The system enters an entropy-positive state: consuming energy without maintaining structural coherence.
In reaction, the cell activates emergency metabolic rewiring to contain disorder. AMPK suppresses ATP-expensive processes while enhancing glycolysis, autophagy, and redox buffering. Glycogen stores are mobilized, and ROS detoxification escalates. This phase buys time through inefficient but essential survival metabolism.
As stress recedes, the system initiates structural and metabolic reprogramming. Mitochondrial function is restored via biogenesis and mitophagy, while fatty acid oxidation assumes a greater role in ATP supply. Glycolysis is normalized, and the redox state is rebalanced. The system transitions from compensation to regeneration.
A new dynamic equilibrium emerges—thermodynamically sustainable and structurally reorganized. Energy production and use are re-coupled; fuel selection becomes context-sensitive; and gene expression, organelle distribution, and cofactor pools reflect embedded stress memory. The cell achieves resilience, not by returning to baseline, but by evolving its metabolic logic.
This phase-based model reveals metabolism as a non-equilibrium choreography of energy and entropy, where cellular coherence is sustained through anticipatory adaptation, not static control.
References
Friston, K. (2010).
The free-energy principle: a unified brain theory?
Nature Reviews Neuroscience, 11(2), 127–138.
https://doi.org/10.1038/nrn2787
PGC-1α integrates the mitochondrial biogenesis and oxidative metabolism programs.
Cell, 127(6), 1109–1122.
https://doi.org/10.1016/j.cell.2006.11.013
Wallace, D. C. (2005).
A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine.
Annual Review of Genetics, 39, 359–407.
https://doi.org/10.1146/annurev.genet.39.110304.095751
Lopez-Lluch, G., et al. (2006).
Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency.
Proceedings of the National Academy of Sciences, 103(6), 1768–1773.
https://doi.org/10.1073/pnas.0510452103
Grahame Hardie, D. (2011).
AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function.
Genes & Development, 25(18), 1895–1908.
https://doi.org/10.1101/gad.17420111